Confermata la peggiore atrocità nella storia del mondo senza alcuna responsabilità o giustizia in arrivo
20 APRILE 2023
Fonte: https://rwmalonemd.substack.com/p/the-worst-atrocity-in-the-history
L'Organizzazione Mondiale della Sanità stima che (in tutto il mondo) ci siano stati 763.740.140 casi confermati di COVID-19, compresi 6.908.554 decessi al 19 aprile 2023. Questo dato non include le componenti aggiuntive dell'eccesso di mortalità durante la crisi COVID documentata da molti nei Paesi occidentali, per la quale gli scienziati e i vari governi sembrano non sapere quale sia l'agente causale e nessun governo sembra voler indagare... Anche se la maggior parte concorderà in privato sul fatto che queste morti sono anche legate alle politiche di "salute pubblica" della COVID-19 in un modo o nell'altro. Queste includono i decessi dovuti alle chiusure (carestie, suicidi, violenza, abuso di alcol e droghe), ai lunghi periodi di COVID, ai decessi dovuti ai vaccini, alla mancanza di cure mediche per il cancro e altre malattie, ecc. Complessivamente, la stima dei decessi totali dovuti alla crisi COVID è probabilmente di circa dieci milioni di persone o più. Dieci milioni di persone sono un numero molto grande. È difficile anche solo da immaginare.
Per fare un paragone, il più grande disastro naturale (escluse le carestie) del XX secolo è stata l'inondazione del fiume Yangtze in Cina nel 1931, che ha ucciso 3,7 milioni di persone sia direttamente che indirettamente, e molte persone sono morte a causa delle scarse condizioni igieniche e delle malattie. Nel 1958, l'alluvione del fiume Giallo in Cina uccise circa un milione di persone, anche se le stime variano molto. Altre inondazioni, cicloni e terremoti hanno ucciso innumerevoli persone. Ma nessuno lo ha fatto con tanta devastazione della vita umana come il virus SARS-CoV-2-WIV.
Ma sappiamo anche che non si è trattato di un disastro naturale, bensì di un disastro provocato dall'uomo.
Un elenco di genocidi su Wikipedia mostra che non ci sono state singole atrocità umane nella storia dell'umanità che si siano avvicinate alle morti causate dalla crisi COVID.
Come facciamo a "saperlo"? Perché abbiamo le ricevute grazie a Judicial Watch e alle indagini del Congresso, tuttora in corso.
Questa settimana, Judicial Watch ha ricevuto 552 pagine dal Dipartimento della Salute e dei Servizi Umani degli Stati Uniti (HHS). Questi documenti comprendono la richiesta iniziale di sovvenzione, i bozzetti, i bilanci e le relazioni annuali della EcoHealth Alliance al NIH. Descrivono gli obiettivi specifici del progetto, che comprendono la creazione di virus mutanti della SARS (e della MERS) "per prevedere meglio la capacità dei nostri CoV [coronavirus] di infettare le persone".
Ho passato il pomeriggio a leggere questi documenti e le 552 pagine sono una miniera d'oro di informazioni. Ma l'obiettivo specifico 3 del contratto è particolarmente importante. Si legge per intero:
Obiettivo specifico 3: testare le previsioni sulla trasmissione interspecie del CoV. Verificheremo i nostri modelli di gamma di ospiti (cioè il potenziale di emergenza) sperimentalmente utilizzando la genetica inversa, saggi di legame di pseudovirus e recettori, ed esperimenti di infezione virale in colture cellulari e topi umanizzati. Con i bat-CoV che abbiamo isolato o sequenziato e utilizzando l'infezione con virus vivi o pseudovirus in cellule di origine diversa o che esprimono molecole recettoriali diverse, valuteremo il potenziale di diffusione di ciascun virus isolato e di quelli con la sequenza del sito di legame del recettore. A tale scopo sequenzieremo i geni delle proteine spike (o di altri recettori che si legano/diffondono) di tutti i nostri bat-CoV, creando mutanti per identificare quanto significativamente ciascuno di essi dovrebbe evolversi per utilizzare ACE2, CD26/DPP4 (recettore del MERS-CoV) o altri potenziali recettori dei CoV. Utilizzeremo quindi saggi di legame tra recettori e pseudovirus mutanti, studi in vitro in linee cellulari di pipistrelli, primati, esseri umani e altre specie, e con topi umanizzati in cui sono stati identificati filogeneticamente o isolati virus particolarmente interessanti. Questi test forniranno dati rilevanti per la salute pubblica e miglioreranno iterativamente il nostro modello predittivo per indirizzare meglio le specie di pipistrelli e i CoV durante i nostri studi sul campo per ottenere i ceppi di Bat-CoV di maggiore interesse per la comprensione dei meccanismi di trasmissione interspecifica.
In seguito, scrivono (pagina 195):
valuteremo il potenziale di diffusione di ciascun virus isolato e di quelli con la sequenza del sito di legame del recettore. A tale scopo sequenzieremo i geni delle proteine spike (o di altri recettori che si legano/diffondono) di tutti i nostri virus dei pipistrelli, creando mutanti per identificare quanto significativamente ciascuno di essi dovrebbe evolversi per utilizzare ACE2, CD26/DPP4 (recettore del MERS-CoV) o altri potenziali recettori del CoV.
È importante capire che, sebbene queste citazioni siano tecniche e non comprensibili a molti, il nocciolo della questione è che questo progetto era ed è una ricerca sul guadagno di funzione. In contrasto con la testimonianza giurata del Dr. Fauci al Congresso.
È importante tirare fuori queste sezioni che evidenziano la ricerca sul guadagno di funzioni condotta che ha portato alla morte di milioni di persone. Questo è l'unico modo che conosco per far capire agli scienziati, ai tribunali e ai politici che non si tratta di una teoria della cospirazione. È una cosa reale. Che queste morti sono state causate da un omicidio colposo.

L'unica domanda da porsi è: si è trattato di un rilascio accidentale o intenzionale del virus creato dall'uomo? È stato un omicidio colposo o un omicidio?
Secondo le 552 pagine rilasciate, l'Istituto di virologia di Wuhan era così sicuro, c'erano assicurazioni in tal senso e le strutture non sono mai state ispezionate dal governo statunitense. Il rischio di fuga di virus mutanti dal laboratorio non è mai stato nemmeno discusso tra i rischi associati alla conduzione di questa ricerca.
Se era così sicuro, non si deve considerare il rilascio intenzionale di questo virus mutante?
La situazione non fa che peggiorare. Il rapporto dell'anno 2 (2016) afferma chiaramente che l'obiettivo specifico 3 per l'anno 3 è stato ampliato per includere anche la conduzione di ricerche sull'aumento della funzione utilizzando il virus MERS!
Obiettivo specifico 3: verificare le previsioni sulla trasmissione interspecie del CoV. Nell'anno 2 saranno intrapresi i seguenti esperimenti (pag. 197)
-Un clone infettivo di MERS-CoV a lunghezza completa sarà costruito con il metodo della genetica inversa. Utilizzando la sequenza S di diversi virus MERS-correlati identificati dai pipistrelli cinesi, verranno costruiti virus chimerici con il gene S dei coronavirus MERS-correlati dei pipistrelli e il backbone del clone infettivo del MERS-CoV per studiare l'uso del recettore e l'infettività del coronavirus MERS-correlato dei pipistrelli.
Il virus MERS (MERS-CoV) è altamente patogeno. Durante l'epidemia del 2012, si sono verificati circa 2.500 casi noti e 800 decessi. Se questi numeri sono corretti, si tratta di un tasso di mortalità del 31%! Il MERS-CoV non sembrava essere altamente infettivo, a differenza del SARS-CoV-2-WIV (il virus creato da Ralph Baric/EcoHealth/WIV).
Si noti che il passaggio sopra riportato include riferimenti alla creazione di nuovi cloni e al loro collegamento con l'infettività della MERS! Riuscite a immaginare se creassero anche un virus MERS più altamente infettivo, da diffondere in tutto il mondo, come la SARS-CoV-2-WIV? La devastazione sarebbe una cosa mai vista al mondo.
Passiamo al rapporto 2017 (pagina 253):
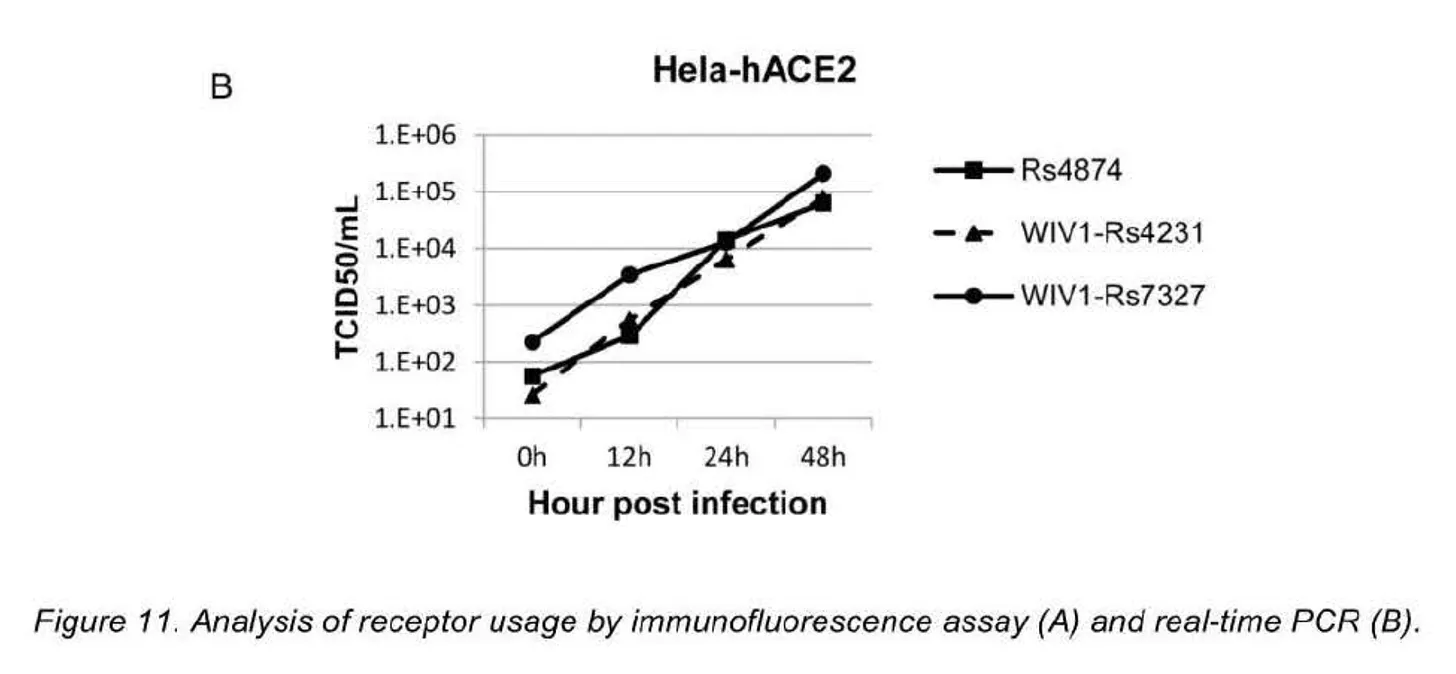
Nell'anno 3, abbiamo isolato con successo Rs4874 dal singolo campione fecale. Utilizzando il sistema di genetica inversa precedentemente sviluppato, abbiamo costruito due virus chimerici con il backbone WIV1 sostituito con il gene S di Rs7327 e Rs4231, rispettivamente. Le cellule Vero E6 sono state infettate rispettivamente con Rs4874, WIV1-Rs4231S e WIV1- Rs7327S e in tutte le infezioni è stata rilevata un'efficiente replicazione del virus mediante saggio di immunofluorescenza. Per valutare l'utilizzo dell'ACE2 umano da parte dei tre nuovi SL-CoV, abbiamo condotto studi sull'infettività del virus utilizzando cellule Hela con o senza l'espressione dell'ACE2 umano. Tutti i virus si sono replicati in modo efficiente nelle cellule con espressione di ACE2 umano. I risultati sono stati ulteriormente confermati dalla quantificazione dell'RNA virale mediante RT-PCR in tempo reale (Fig. 11).
È stato costruito con successo il clone di eDNA infettivo full-length di MERS-CoV. Il gene S completo di 12 nuovi coronavirus legati alla MERS è stato amplificato e clonato nei vettori T. Nel quarto anno, ci proponiamo di utilizzare il metodo della genetica inversa e di costruire virus chimerici con il backbone del MERS-CoV e i geni S di diversi coronavirus legati alla MERS dei pipistrelli recentemente identificati, per esaminare la patogenicità dei coronavirus legati alla MERS dei pipistrelli a livello cellulare e animale.
Più ricerca sul guadagno di funzioni.
Passaggio all'anno 4 (pagina 275):
Obiettivo specifico 3: verificare le previsioni sulla trasmissione interspecifica dei CoV.
Infezione in vivo di topi esprimenti ACE2 umano (hACE2) con varianti della proteina S di SARSr-CoV
Utilizzando i metodi di genetica inversa precedentemente sviluppati, sono stati costruiti cloni infettivi con il backbone WIV1 e la proteina spike di SHC014, W IV16 e Rs4231, rispettivamente, e i virus ricombinanti sono stati salvati con successo. Nell'anno 4, abbiamo eseguito un'infezione preliminare in vivo di SARSr-CoV su topi transgenici che esprimono hACE2. I topi sono stati infettati con 105 pfu di virus ricombinante full-length di WIV1 (rWIV1) e con i tre virus chimerici con punte diverse. La patogenesi dei 4 SARSr-CoV è stata quindi determinata in un ciclo di 2 settimane. I topi infettati con rWIV1-SHC014S hanno subito una perdita di peso corporeo di circa il 20% entro il 6° giorno dall'infezione, mentre WIV1 e rWIV-4231S hanno prodotto una perdita di peso corporeo minore. Nei topi infettati con rWIV1 -WIV16S non è stata osservata alcuna perdita di peso corporeo (Fig. 35a). 2 e 4 giorni dopo l'infezione, la carica virale nei tessuti polmonari dei topi infettati con rWIV1-SHC014S, rWIV1-WIV16S e rWIV1-Rs4231 S ha raggiunto più di 106 copie/g di genoma ed è risultata significativamente superiore a quella dei topi infettati con rWIV1 (Fig. 35b). Questi risultati dimostrano la diversa patogenicità dei SARSr-CoV con diverse proteine spike nei topi umanizzati.
Nel 2020, sembra che il contratto sia stato rivisto e prorogato per altri CINQUE anni!

Per questo periodo (2020-2025), sembra che l'obiettivo 3 sulla pagina di copertina sia stato riscritto per eliminare qualsiasi ricerca sul guadagno di funzioni dalla prima pagina della proposta. È come se pensassero di poter essere incolpati di aver condotto una ricerca sul guadagno di funzioni che ha portato allo sviluppo di un virus che è stato rilasciato sulla popolazione mondiale! Sul serio, la completa riscrittura dell'obiettivo 3 sulla pagina di copertina del nuovo contratto, per rimuovere ogni allusione alla creazione di virus mutanti, ha l'aspetto di un insabbiamento di una delle atrocità più letali al mondo.
Obiettivo 3. Caratterizzazione in vitro e in vivo del rischio di spillover della SARSr-CoV, unita ad analisi spaziali e filogenetiche per identificare le regioni e i virus che destano preoccupazione per la salute pubblica. Utilizzeremo i dati di sequenza della proteina S, la tecnologia dei cloni infettivi, gli esperimenti di infezione in vitro e in vivo e l'analisi del legame con i recettori per verificare l'ipotesi che le soglie di divergenza % nelle sequenze della proteina S predicano il potenziale di spillover. Combineremo questi dati con la distribuzione dell'ospite pipistrello, la diversità e la filogenesi virale, l'indagine umana sui comportamenti a rischio e sulla malattia e la sierologia per identificare i punti caldi del rischio di spillover della SARSr-CoV nella Cina meridionale. L'insieme di questi dati e analisi sarà fondamentale per lo sviluppo futuro di interventi di salute pubblica e di una maggiore sorveglianza per prevenire la ricomparsa della SARS o la comparsa di un nuovo SARSr-CoV.
È interessante notare che, più in profondità nel testo, la proposta è un po' più specifica sull'obiettivo 3.
Obiettivo 3: caratterizzazione in vitro e in vivo del rischio di spillover della SARSr-CoV, insieme ad analisi spaziali e filogenetiche per identificare le regioni e i virus che destano preoccupazione per la salute pubblica. Caratterizzeremo la propensione dei nuovi SARSr-CoV a infettare le persone in vitro utilizzando cellule epiteliali primarie delle vie aeree umane e in vivo utilizzando il modello murino transgenico hACE2. Utilizzeremo trattamenti con mAb e vaccini per verificare la nostra ipotesi che i SARSr-CoV con una divergenza del 10-25% nelle sequenze della proteina S rispetto al SARS-CoV sono probabilmente in grado di infettare le cellule umane e di eludere i farmaci mAb e i vaccini. In seguito, mapperemo la distribuzione geografica dei loro ospiti pipistrelli e altri fattori di rischio ecologico per identificare i principali "punti caldi" di rischio per future ricadute.
Si noti l'uso della parola "nuovo". Non è chiaro se questi nuovi mutanti siano già stati "sviluppati" (ricerca sul guadagno di funzione) negli anni precedenti o se debbano essere sviluppati.
Più avanti nei documenti, si legge (pagina 496):
3.3.a Costruzione di virus chimerici SARSr-CoV: Infettare i cloni con il gene S dei nuovi SARSr-CoV e il backbone del genoma del SARSr-CoV WIV1 utilizzando il sistema di genetica inversa sviluppato nella nostra precedente R01 (24). I cloni BAC infettivi corretti saranno esaminati mediante digestione del DNA BAC con un enzima di restrizione appropriato o amplificazione PCR. I virus chimerici saranno salvati in cellule Vero e poi verificati mediante analisi di sequenza.
La proposta prosegue descrivendo come i virus chimerici infetteranno cellule epiteliali primarie e topi umanizzati (pagine 496-497).
Già! Non è cambiato nulla. In fondo al testo c'è la ricerca sul guadagno di funzione che devono ancora fare! È stata solo rimossa dalla prima pagina della proposta.
Non ci sono più rapporti annuali, quindi qualsiasi ricerca sia stata condotta successivamente non è nota dopo il rapporto annuale del 2019.
Questa ricerca deve cessare ora. Il Congresso deve bloccare immediatamente i finanziamenti. Deve esserci responsabilità. Deve esserci giustizia per i feriti e i morti.
Ci sono dieci milioni di persone morte a causa di questo "progetto" di ricerca. Abbiamo bisogno di un'altra epidemia causata dall'uomo per comprendere appieno quanto sia pericoloso questo tipo di ricerca?